2.1. Input file
DiveR requires either aligned sequence file(s) or DiMA output file(s) (JSON format) as input file(s), where DiveR will convert and concatenate them (the inputs) into a single CSV file (Figure 2), which will act as the source for subsequent data visualisation. Each aligned sequence / DiMA output file is treated as one viral protein. Currently, DiveR accepts aligned FASTA or JSON files generated using multiple sequence alignment (MSA) tools and DiMA, respectively.
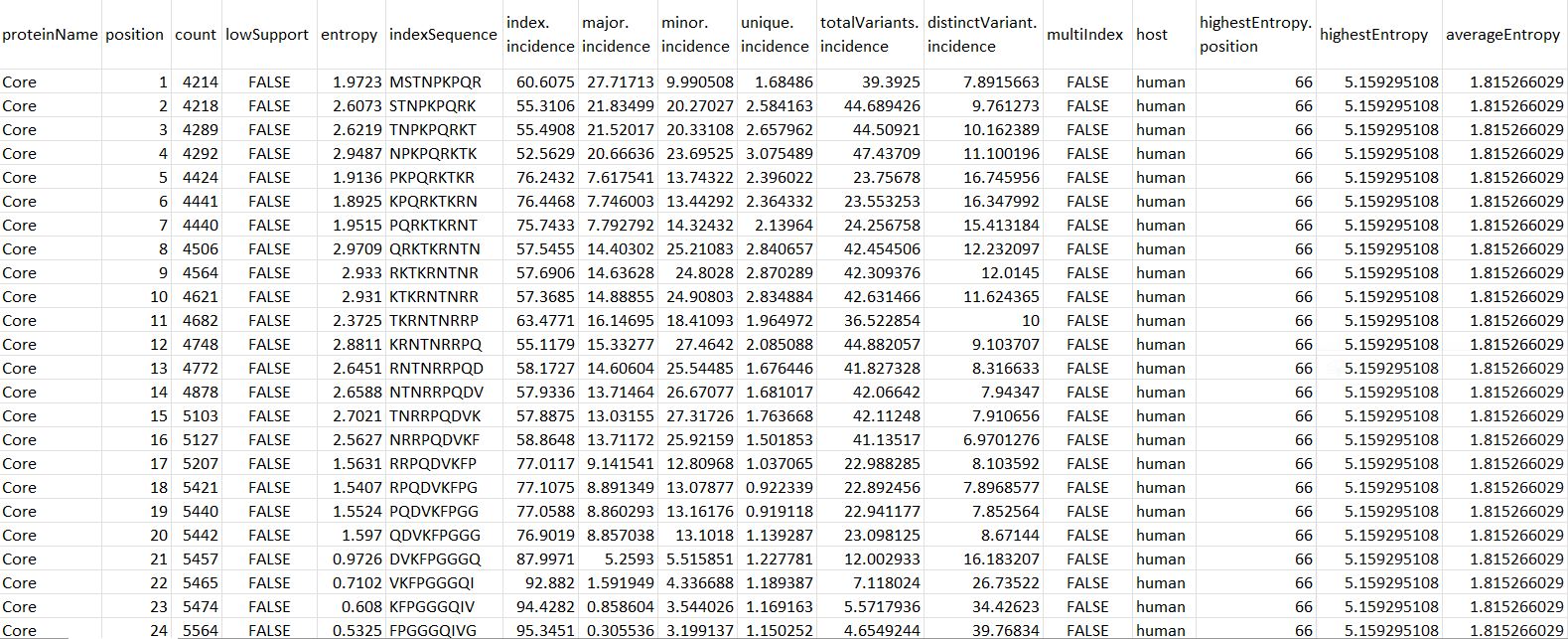
Figure 2. DiMA JSON-Converted CSV Output Format.
proteinName: name of the protein
position: starting position of the aligned, overlapping k-mer window
count: number of k-mer sequences at the given position
lowSupport: k-mer position with sequences lesser than the minimum support threshold (TRUE) are considered of low support, in terms of sample size
entropy: level of variability at the k-mer position, with zero representing completely conserved
indexSequence: the predominant sequence (index motif) at the given k-mer position
index.incidence: the fraction (in percentage) of the index sequences at the k-mer position
major.incidence: the fraction (in percentage) of the major sequence (the predominant variant to the index) at the k-mer position
minor.incidence: the fraction (in percentage) of minor sequences (of frequency lesser than the major variant, but not singletons) at the k-mer position
unique.incidence: the fraction (in percentage) of unique sequences (singletons, observed only once) at the k-mer position
totalVariants.incidence: the fraction (in percentage) of sequences at the k-mer position that are variants to the index (includes: major, minor and unique variants)
distinctVariant.incidence: incidence of the distinct k-mer peptides at the k-mer position
multiIndex: presence of more than one index sequence of equal incidence
host: species name of the organism host to the virus
highestEntropy.position: k-mer position that has the highest entropy value
highestEntropy: highest entropy values observed in the studied protein
averageEntropy: average entropy values across all the k-mer positions
2.2. Parameters
2.2.1. Input Parameters
2.2.1.1. Host Name
Species name of the organism host to the studied virus.
2.2.1.2. Size of k-mer
k-mer, a window with size of k, gives us the overview, overall diversity of that particular window. By default, DiMA uses k-mer size of nine to evaluate the viral diversity, with respect to cellular immune response.
2.2.1.3. Protein Name
Name of the protein.
2.2.1.4. Support Threshold
Support is defined as the number of sequences at a given k-mer position that are free of gaps, unknown or ambiguous nucleotide bases, and amino acid residues. Positions with less than 30 sequences (default) are defined as of low support.
2.2.1.5. Sequence Type
Nucleotide or amino acid sequence.
2.2.2. Display Parameters
2.2.2.1. Host Number Selection
Select the number of host studied (one (default) or two hosts). DiveR supports co-visualization of viral diversity dynamics between two hosts.
2.2.2.2. Font Size
Font size displayed on the plots.
2.2.2.3. Line and Dot Size
Line and dot size displayed on the plots.
2.2.2.4. Protein Names in Order
Determine the order of proteins displayed on plot (Please ensure the protein names provided are the same as the one used in input run!).